I am currently a software developer at Stanford University working for the Markland research group in the Department of Chemistry. Here, I am part of a team working on the OpenMM molecular dynamics simulation toolkit.
I was previously a graduate student at the University of California, Santa Barbara in the Department of Chemical Engineering and was a member of the Shell research group. My research interests during that time were related to molecular simulation and multiscale modeling: in particular, coarse-grained modeling and coarse-graining method development, with a focus on applications for the study amyloid aggregation of peptides. Before this, I was an undergraduate student at Lehigh University in the Department of Chemical and Biomolecular Engineering, and worked in the Mittal research group (now at Texas A&M University). There, I performed research studying colloidal self-assembly using molecular dynamics simulations, and developing tools for colloidal crystal structure prediction.
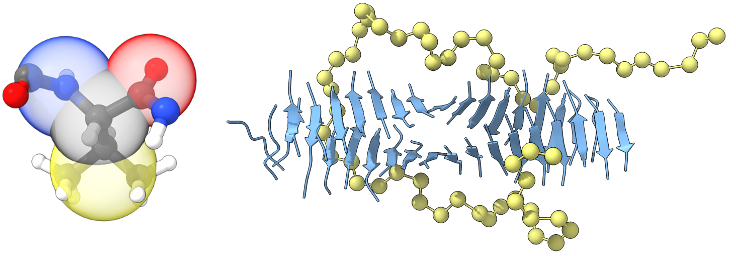
Multiscale approaches are crucial for modeling various phenomena in molecular systems that involve length and time scales out of reach of detailed atomistic simulations. My current work in the Shell group is broadly focused on applying the relative entropy coarse-graining method to address various multiscale modeling challenges. Building coarse-grained models using systematic bottom-up approaches such as relative entropy minimization requires only that information from detailed atomistic simulations of systems of interest be available: all model parameters can be optimized together, and no ad hoc tuning is necessary in principle to accurately recover configurational properties of the underlying detailed systems.
A coarse-grained model should not only accurately represent the underlying properties of the reference systems that were used to build it, however; in practice, it is also important that a useful coarse-grained model should be transferable to new molecules, mixtures, and thermodynamic conditions outside of its reference set. I have implemented and validated a microcanonical coarse-graining approach [11] to improve the temperature transferability of relative entropy coarse-grained models by embedding information about energy fluctuations that would ordinarily be lost during coarse-graining. More recently, I have been applying coarse-graining to peptide systems to study aggregation. Tau protein aggregation is implicated in Alzheimer's disease and much remains unknown about the molecular mechanisms underlying tau fibril formation. A coarse-grained model of the PHF6 motif [12] (found in the tau protein and known to be crucial for its aggregation) enables large-scale simulations of fibril growth and can shed light on processes involved in amyloid aggregation. Presently, I am extending the modeling methodology that we have applied for PHF6 to develop transferable bottom-up coarse-grained peptide models over the broader sequence space of a reduced alphabet of amino acids.
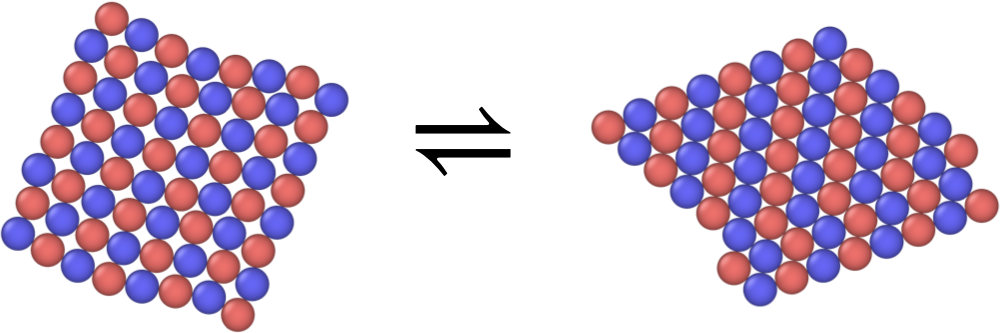
Tuning the self-assembly of colloidal particles into ordered crystalline lattices has many applications for material design and also serves as a model problem for understanding some of the fundamental physics behind crystal nucleation and growth. Systems of DNA-functionalized particles in particular, in which different proportions of different pairs of complementary DNA strands can be grafted to the surfaces of different types, sizes, and shapes of particles, effectively allow control over the strengths and ranges of all of the pairwise interactions in a multicomponent system [1][3]. In my work in the Mittal group, I used molecular simulation techniques to probe the relationships between interactions in these systems, the resulting thermodynamics and kinetics of self-assembly, and how they lead to the selection of particular ordered structures during crystallization. In particular, we have investigated the influence of finite system size on crystal structure preference, showing that structural transformations observable during self-assembly of certain binary colloidal mixtures can be explained thermodynamically [5][10] (we also have a software package PyPhase implementing an extension to finite and semi-periodic geometries of the Frenkel-Ladd method for solid free energies [4]). We have also shown that symmetry groups can be employed to efficiently explore the space of possible structures into which a mixture of colloids can crystallize, and to predict multicomponent phase diagrams for such systems [2][6] (the paccs software package [7] is available to perform such computations for two-dimensional colloidal crystals).
Copyright ©2017–2024 Evan Pretti. All rights reserved.